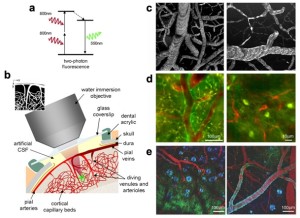
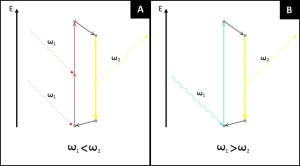
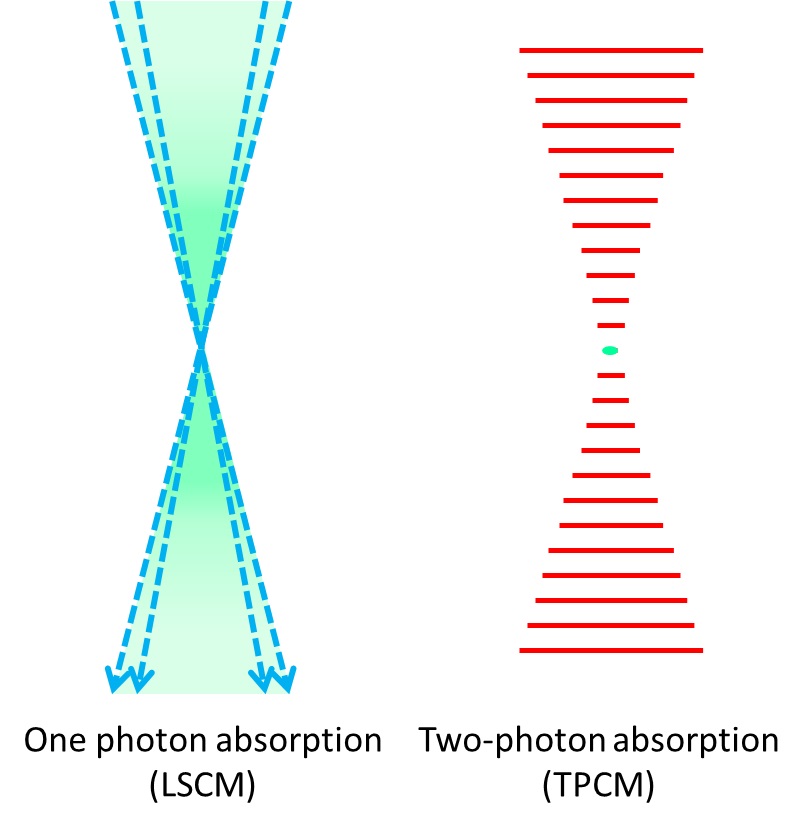
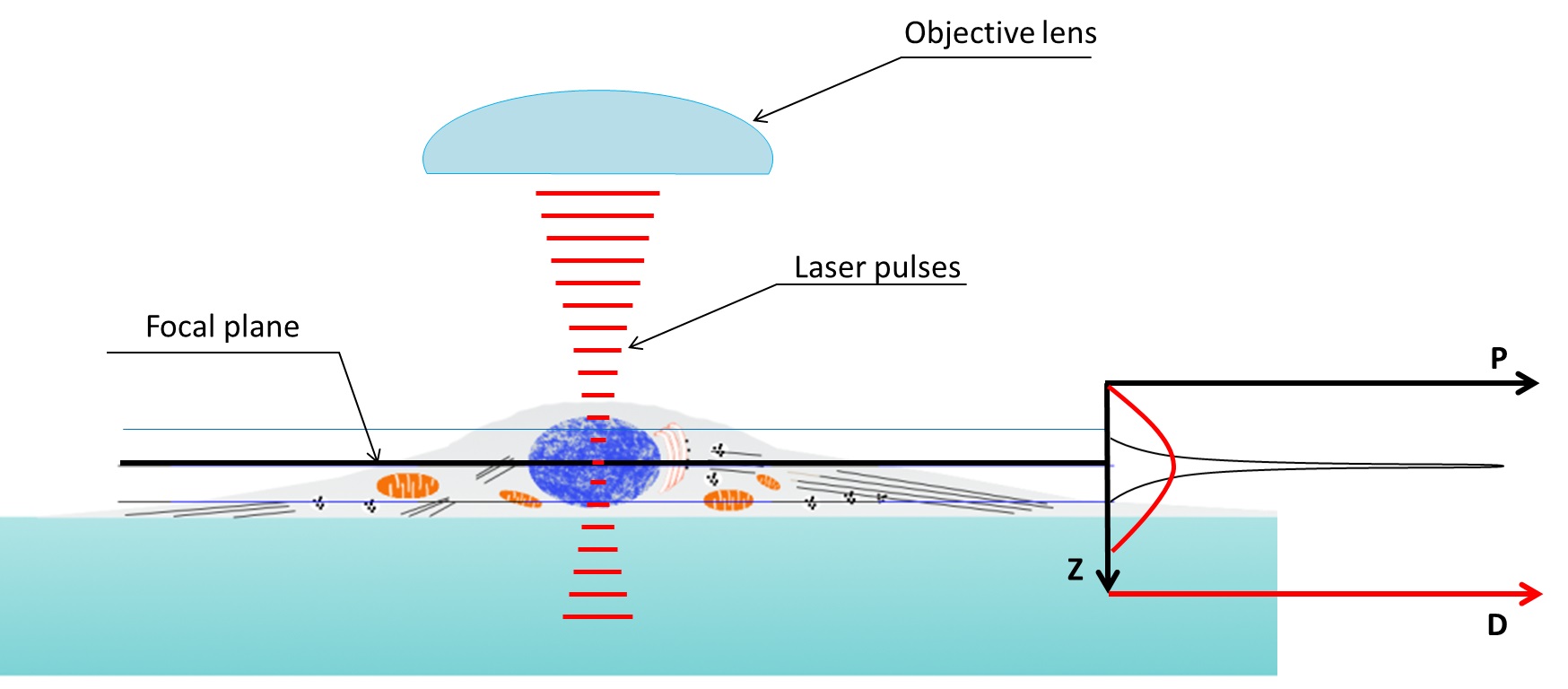
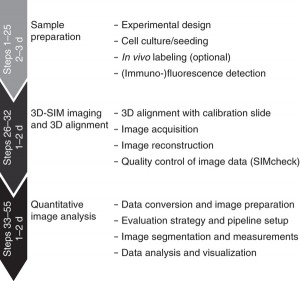
 Super Resolution Microscopy: The Diffraction Limit of Light...
Super Resolution Microscopy: The Diffraction Limit of Light...Introduction to Diffraction Limit of Light The optical microscopy, also called the light microscopy, is the oldest technique of microscopy w...
Read more Wide-field fluorescence microscopy (Fluorescent microscopy)...
Wide-field fluorescence microscopy (Fluorescent microscopy)...Introduction to fluorescence microscopy Fluorescence is a natural phenomenon in which following the absorption of light by a molecule (fluor...
Read more CherryTemp System on GE Delta Vision Elite and GE Delta Vision OM...
CherryTemp System on GE Delta Vision Elite and GE Delta Vision OM...CherryTemp System on GE Delta Vision Elite and GE Delta Vision OMX V4 and possible ways to mount the system on customized microscope systems...
Read more